The HYAK team has taken six concrete steps to stabilize and optimize storage on KLONE over the past few weeks.
While the storage on KLONE (i.e., mmfs1 or gscratch) may appear to be a monolithic device, it is an extremely complex cluster in its own right. This storage cluster is mounted on every KLONE node: so despite appearing as "on the node", gscratch physically resides on specialized storage hardware separated from the compute resources of KLONE. The storage is accessed across a high-speed, ultra low-latency HDR Infiniband network, and is designed to be scalable independent of KLONE’s compute resources.
As mentioned in an earlier blog post today, our incoming hardware expansion will drastically increase the amount of demand the storage cluster can handle. In the meantime, the Hyak team has taken measures to help maintain a usable level of storage performance for users and jobs:
1. Improved internal storage metrics gathering and visibility.#
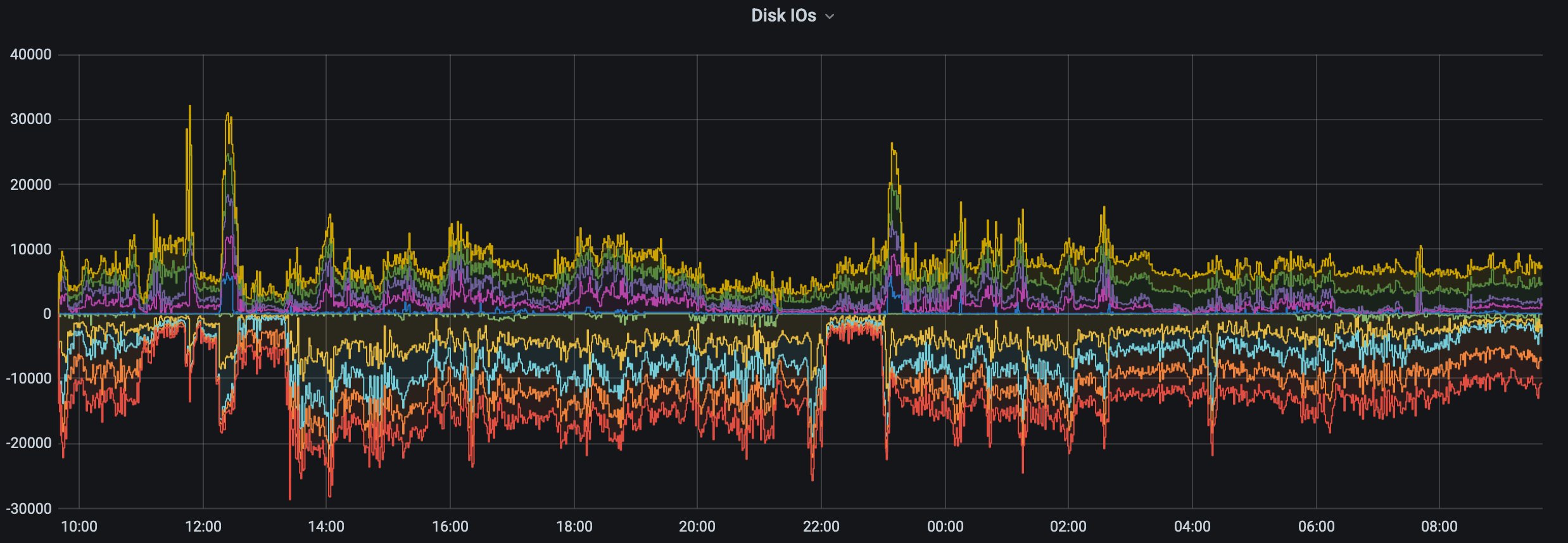
The HYAK team improved storage-cluster metric gathering and visibility, allowing us to correlate those metrics to reports of poor user experience, and to make data-driven tuning and storage policy decisions.
In the figure above we have visibility into if an abnormally high number of jobs have errors that might suggest underlying storage or other user experience issues.
2. Created custom filesystem migration policies to optimize the use of the NVMe layer.#
The bulk of the storage capacity on KLONE is stored on rotary hard disk drives totalling approximately 1.7 Petabytes (PB) of raw storage. In addition to the hard disk storage, there is a much smaller, extremely fast–and expensive–pool of NVMe "flash" storage that functions both as a write buffer for new files written to the filesystem, and also as a read-cache-like layer where files can be read without causing load on the rotary disks.
The HYAK team has also optimized the file placement policy: files most likely to generate heavy load reside in the limited space of the NVMe layer, ensuring that no storage load is generated on the hard disk layer when those files are repeatedly accessed.
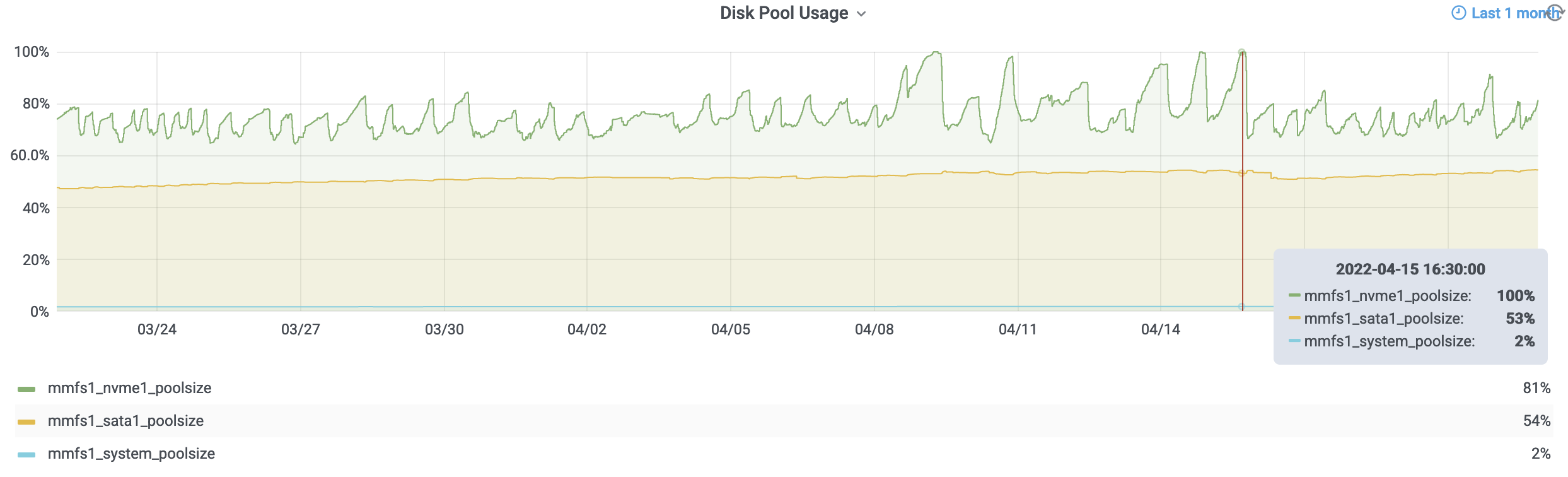
In the figure above you can see that the flash tier (green line) is allowed to fill up to 80% capacity due to job writes then the migration policy begins until the flash tier is down to 65% full. For the majority of the past few several weeks we can see things worked as expected. However, there were a few events recently where jobs were producing so much data that the flash tier was able to get to 100% full faster than the storage system could move data off the flash tier. Giving the migration process too high of a priority results in "slowness" in the user experience. We have since been tuning the aggressiveness of this migration process to reduce the likelihood of it occuring again.
3. Added QoS policies to improve worst-case filesystem responsiveness.#
The KLONE filesystem has a coarse Quality-of-Service (QoS) tuning facility that allows the filesystem to cap the rate of storage operations for various types of storage input-output (IO). The HYAK team has used this facility in two different ways:
First, to limit the storage load impact when the NVMe layer, described above, needs to free up space by moving files to the hard drive layer.
Secondly, to moderate the amount of storage load that can be generated by any single compute node in the cluster. This way, outlier jobs in terms of storage load generation are less likely to have an outsized performance impact on the storage.
4. Manually identifying jobs causing a disproportionate impact on storage performance.#
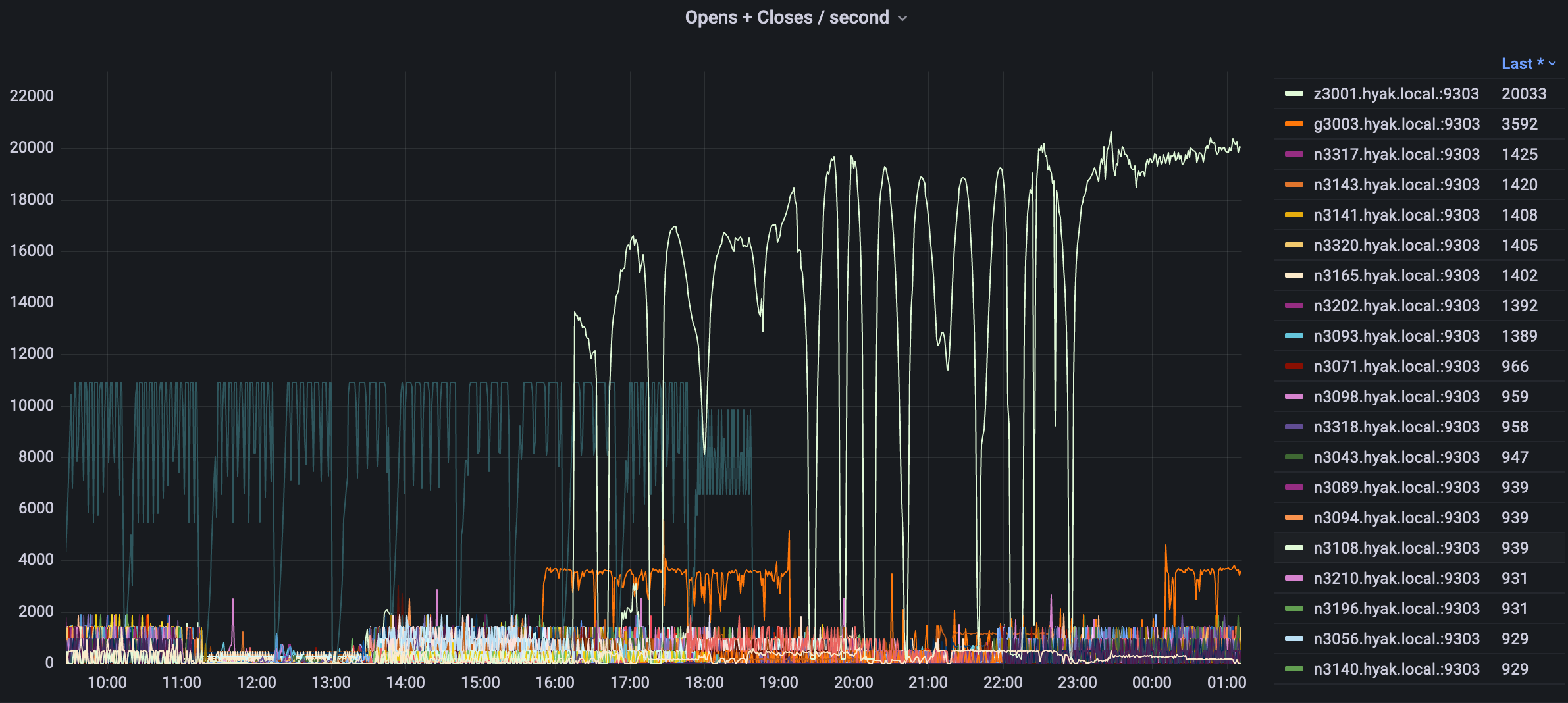
Utilizing metrics and old-fashioned sleuthing, we have been manually tracking down individual jobs that appear to be having a disproportionate and/or unnecessary impact on storage performance, and working with users to address the storage performance impact of these jobs.
In the above figure we can see job IO follows a power law dynamic, a small handful of jobs are often responsible for the majority of load. In this case a single job on a single node is responsible. When users report storage "slowness" this disrepancy can be even more pronounced but we are able to quickly narrow down which specific nodes are responsible and address these corner cases.
5. Dynamically reducing the number of running checkpoint partition jobs.#
As of April 19th, 2022, we have implemented data-driven automation to moderate storage load by dynamically managing the number of running checkpoint (ckpt) partition jobs. When the number of running ckpt jobs is being limited, pending jobs will show AssocGrpJobsLimit as the REASON for not starting.
Please note that non-ckpt jobs (i.e., jobs submitted to nodes your lab contributed to the cluster) are not limited in any way. The social contract when joining the HYAK community is that you get access to the nodes your lab contributes on-demand, and–if and when they are idle–access to other labs’ resources on the cluster. However, access to other labs’ resources isn’t and hasn’t ever been guaranteed: it’s just that there’s often a steady state idle capacity for users to "burst" into by submitting ckpt jobs.
In aggregate, 'Storage Load' is a consumable resource just like CPU cores or memory, albeit one that impacts the whole cluster when it is over-consumed. The SLURM cluster scheduler cannot directly consider storage load availability when evaluating resources for starting ckpt jobs, hence our need to automate. Our new tooling limits the storage performance impact from ckpt jobs in order to improve storage stability for everyone.
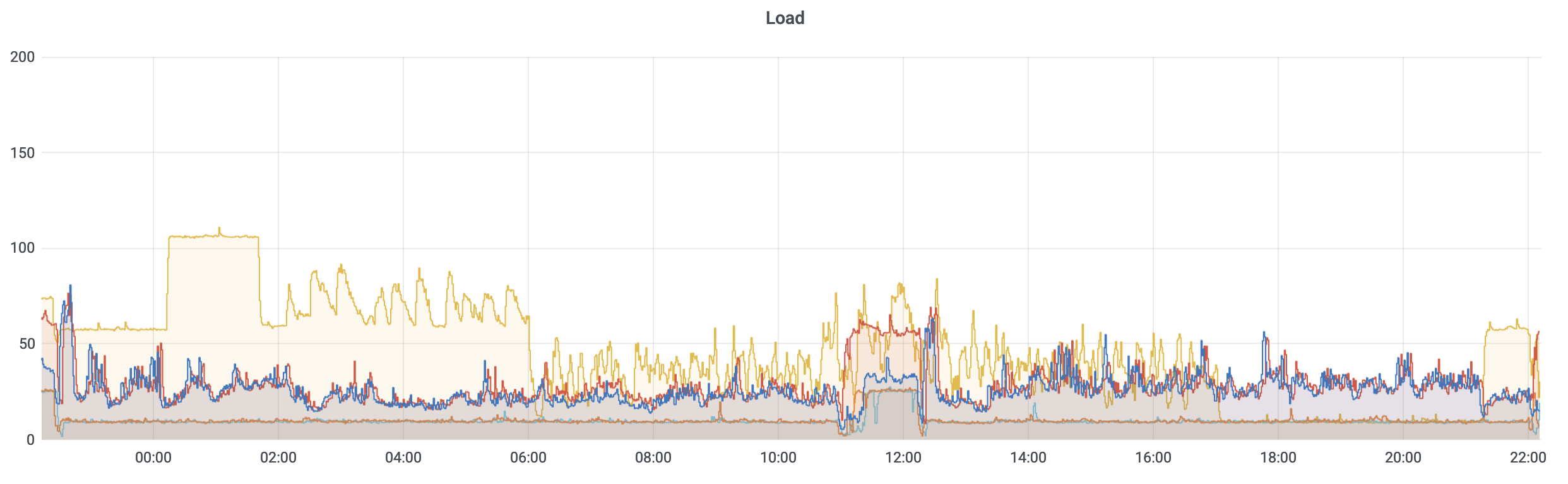
The red and blue lines represent two storage servers that we have most closely tied to the user experience and 50% load being the threshold we aim to remain at or under by dynamically reducing the number of running ckpt jobs when it exceeds that limit.
So far, this appears to be very effective at moderating the overall storage load, preventing the storage cluster from becoming unusably slow and avoiding other storage-performance issues. We will continue to tune it in search of the best balance between idle resource utilization via ckpt and storage performance.
6. Expanding the team#
Acknowledging that the storage sub-system is a complicated machine in its own right, it needs much more care and attention and the current HYAK team is stretched incredibly thin as is. We have started the process of hiring a dedicated research data storage systems engineer to focus on optimizing storage going forward.
See also:




