JuiceFS or using Kopah on Klone

If you haven't heard, we recently launched an on-campus S3-compatible object storage service called Kopah docs that is available to the research community at the University of Washington. Kopah is built on top of Ceph and is designed to be a low-cost, high-performance storage solution for data-intensive research.
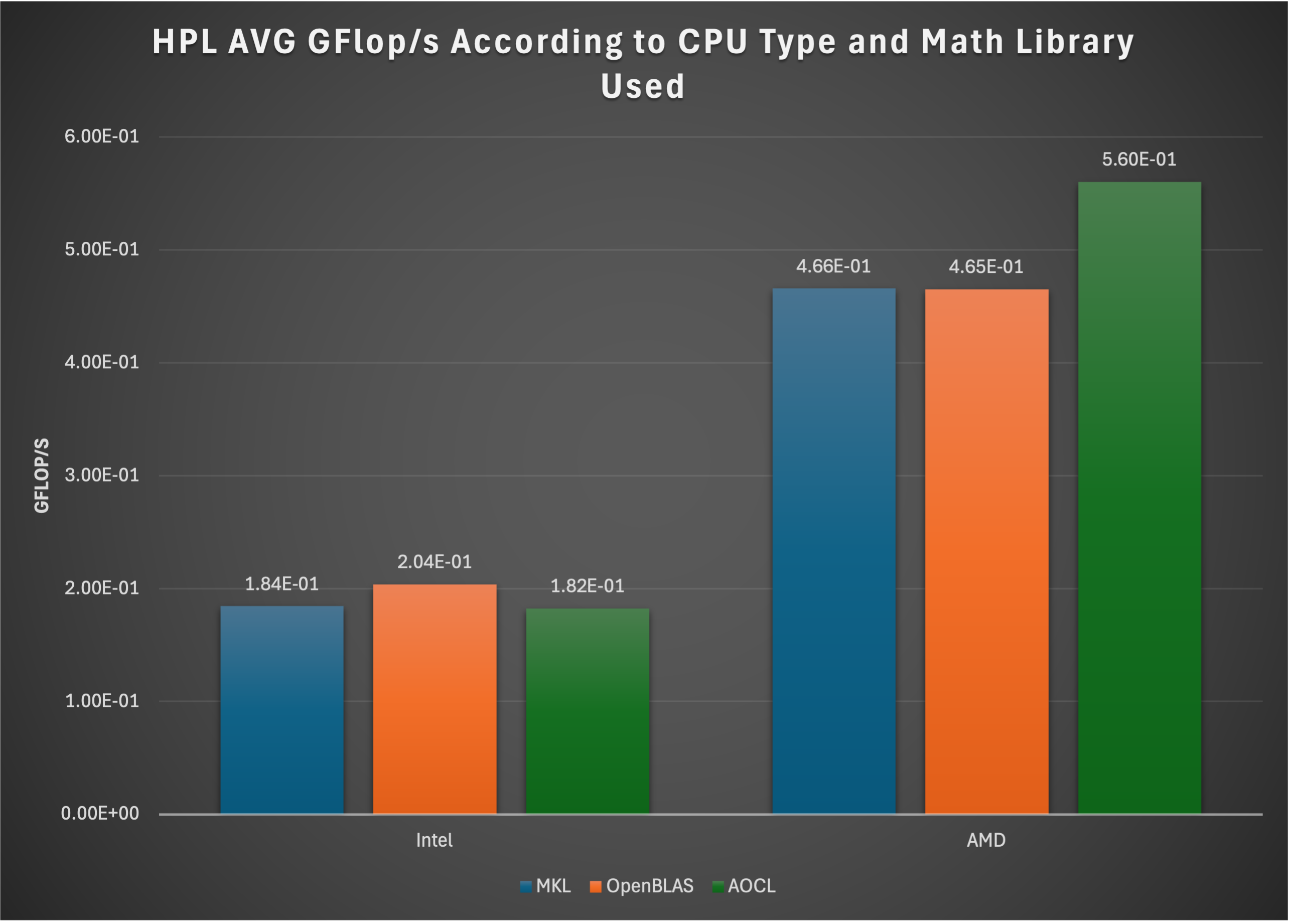
From our testing we have observed significant performance challenges for JuiceFS in "single mode" which is demonstrated by this blog post. We do not recommend JuiceFS as a solution for demanding workflows.
While the deployment of Kopah was welcome news to those who are comfortable working with S3-compatible cloud solutions, we recognize some folks may be hesitant to give up their familiarity with POSIX file systems. If that sounds like you, we explored the use of JuiceFS, a distributed file system that provides a POSIX interface on top of object storage, as a potential solution.
Simplistically, object storage often presents using two API keys and data is accessed using a command line tool that wraps API calls, whereas POSIX is what you typically get presented with from the storage when interacting with a cluster via command-line.
Installation
JuiceFS isn't installed by default so you will need to compile it yourself or download the pre-compiled binary from their release page.
As of January 2025 the latest version is 1.2.3 and you want the amd64 version if using from Klone. The command below will download and extract the binary to your current working directory.
wget https://github.com/juicedata/juicefs/releases/download/v1.2.3/juicefs-1.2.3-linux-amd64.tar.gz -O - | tar xzvf -
I have to move it to a folder in my $PATH so I can run it from anywhere by just calling the binary. Your personal environment varies here.
mv -v juicefs ~/bin/
Verify you can run JuiceFS.
npho@klone-login03:~ $ juicefs --version
juicefs version 1.2.3+2025-01-22.4f2aba8
npho@klone-login03:~ $
Cool, now we can start using JuiceFS!
Standalone Mode
There are two ways to run JuiceFS, standalone or distributed mode. This blog post explores the former. Standalone mode is meant to only present Kopah via POSIX on Klone. The key points being:
- There is an active
juicefsprocess required to run while you want to access it. - It is intended for you to run it only on the node you are running the process from.
If you wanted to run JuiceFS on multiple nodes or with multiple users then we will have another proof-of-concept with distributed mode in the future.
Create Filesystem
JuiceFS separates the data (placed into S3 object storage) and the metadata, which is kept locally in a database. The command below will create the myjfs filesystem and store the metadata in a SQLite database called myjfs.db in the directory where the command is run. It puts the data itself into a Kopah bucket called npho-project.
juicefs format \
--storage s3 \
--bucket https://s3.kopah.uw.edu/npho-project \
--access-key REDACTED \
--secret-key REDACTED \
sqlite3://myjfs.db myjfs
You can rename the metadata file and the filesystem name to whatever you want (they don't have to match). The same goes for the bucket name on Kopah. However, I would strongly recommend having unique metadata file names that match the file system names for ease of tracking alongside the bucket name itself.
npho@klone-login03:~ $ juicefs format \
> --storage s3 \
> --bucket https://s3.kopah.uw.edu/npho-project \
> --access-key REDACTED \
> --secret-key REDACTED \
> sqlite3://myjfs.db myjfs
2025/01/31 11:52:47.940709 juicefs[1668088] <INFO>: Meta address: sqlite3://myjfs.db [interface.go:504]
2025/01/31 11:52:47.944930 juicefs[1668088] <INFO>: Data use s3://npho-project/myjfs/ [format.go:484]
2025/01/31 11:52:48.666657 juicefs[1668088] <INFO>: Volume is formatted as {
"Name": "myjfs",
"UUID": "eb47ec30-c1f7-4a92-9b17-23c4beae7f76",
"Storage": "s3",
"Bucket": "https://s3.kopah.uw.edu/npho-project",
"AccessKey": "removed",
"SecretKey": "removed",
"BlockSize": 4096,
"Compression": "none",
"EncryptAlgo": "aes256gcm-rsa",
"KeyEncrypted": true,
"TrashDays": 1,
"MetaVersion": 1,
"MinClientVersion": "1.1.0-A",
"DirStats": true,
"EnableACL": false
} [format.go:521]
npho@klone-login03:~ $
You can verify there is now a myjfs.db file in your current working directory. It's a SQLite database file that will store your file system meta data.
We can also verify the npho-project bucket was created on Kopah to store the data itself.
npho@klone-login03:~ $ s3cmd -c ~/.s3cfg-default ls
2025-01-31 19:48 s3://npho-project
npho@klone-login03:~ $
You should run juicefs format --help to view the full range of options and customize the parameters of your file system to your unique needs but just briefly:
- Encryption: When you create the file system and format it you can see it has encryption by default using AES256. You can over ride this using the
--encrypt-algoflag if you preferchacha20-rsaor you can use key file based encryption and provide your private key using the--encrypt-rsa-keyflag. - Compression: This is not enabled by default and there is a computational penalty for doing so if you want to access your files since it needs to be de or re encrypted on the fly.
- Quota: By default there is no block (set with
--capacityin GiB units) or inode (set with--inodesfiles) quota enforced at the file system level. If you do not explicitly set this, it will be matched to whatever you get from Kopah. This is still useful for setting explicitly if you wanted to have multiple projects or file systems in JuiceFS that use the same Kopah account and have some level of separation. - Trash: By default, files are not deleted immediately but moved to a trash folder similar to most desktop systems. This is set with the
--trash-daysflag and you can set it to0if you want files to be deleted immediately. The default here is 1 day after which the file is permanently deleted.
Mount Filesystem
Running the command below will mount your newly created file system to the myproject folder in your home directory. It does not need to previously exist.
juicefs mount sqlite3://myjfs.db ~/myproject --background
The SQLite database file is critical, do not lose it. You can move its location around afterwards but it contains all the meta data about your files.
This process occurs in the background.
Where you mount your file system the first time is where it will be expected to be mounted going forward.
npho@klone-login03:~ $ juicefs mount sqlite3://myjfs.db ~/myproject --background
2025/01/31 11:57:01.652279 juicefs[1690855] <INFO>: Meta address: sqlite3://myjfs.db [interface.go:504]
2025/01/31 11:57:01.654920 juicefs[1690855] <INFO>: Data use s3://npho-project/myjfs/ [mount.go:629]
2025/01/31 11:57:02.156898 juicefs[1690855] <INFO>: OK, myjfs is ready at /mmfs1/home/npho/myproject [mount_unix.go:200]
npho@klone-login03:~ $
Use Filesystem
Now with the file system mounted (at ~/myproject) you can use it like any other POSIX file system.
npho@klone-login03:~ $ cp -v LICENSE myproject
'LICENSE' -> 'myproject/LICENSE'
npho@klone-login03:~ $ ls myproject
LICENSE
npho@klone-login03:~ $
Remember, you won't be able to see it in the bucket because it is encrypted before being stored there.
Recover Deleted Files
If you enabled the trash can option then you can recover files up until the permanent delete date.
First delete a file on the file system.
npho@klone-login03:~ $ cd myproject
npho@klone-login03:myproject $ rm -v LICENSE
removed 'LICENSE'
npho@klone-login03:myproject $
Verify the file is deleted. Go to recover it from the trash bin.
npho@klone-login03:myproject $ ls
npho@klone-login03:myproject $ ls -alh
total 23K
drwxrwxrwx 2 root root 4.0K Jan 31 12:54 .
drwx------ 48 npho all 8.0K Jan 31 13:08 ..
-r-------- 1 npho all 0 Jan 31 11:57 .accesslog
-r-------- 1 npho all 2.6K Jan 31 11:57 .config
-r--r--r-- 1 npho all 0 Jan 31 11:57 .stats
dr-xr-xr-x 2 root root 0 Jan 31 11:57 .trash
npho@klone-login03:myproject $ ls .trash
2025-01-31-20
npho@klone-login03:myproject $ ls .trash/2025-01-31-20
1-2-LICENSE
npho@klone-login03:myproject $ cp -v .trash/2025-01-31-20/1-2-LICENSE LICENSE
'.trash/2025-01-31-20/1-2-LICENSE' -> 'LICENSE'
npho@klone-login03:myproject $ ls
LICENSE
npho@klone-login03:myproject $
As you can see, we can recover files that are tracked by their delete date. You would need to copy the file back out to recover it.
Unmount Filesystem
When you are done using the file system you can unmount it with the command below.
npho@klone-login03:~ $ juicefs umount myproject
npho@klone-login03:~ $
Remember, the file system is only accessible in standalone mode so long as a juicefs process is running. Since we ran it in the background you will need to explicitly unmount it.
Questions?
Hopefully you found this proof-of-concept useful. If you have any questions for us, please reach out to the team by emailing help@uw.edu with Hyak somewhere in the subject or body. Thanks!